New Drug May Bypass SARS-CoV-2 Blockade Of Innate Immune Response
(Posted on Friday, June 4, 2021)
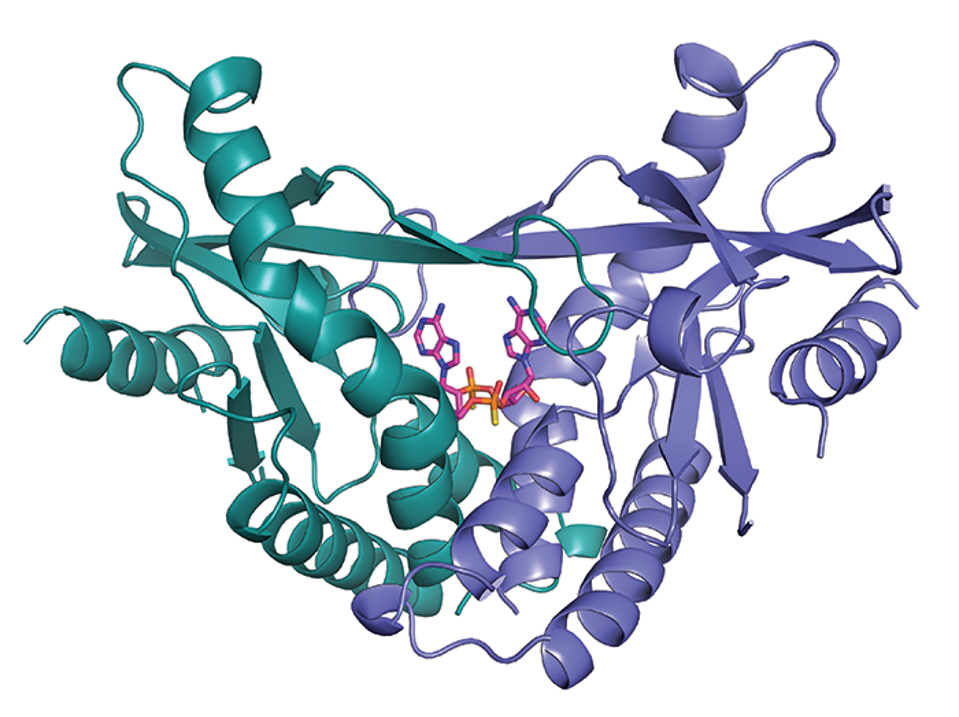
This is the crystal structure of human STING, bond with a synthetic a drug currently in clinical trials for cancer. The same drug stops SARS-Cov-2 from infecting and killing susceptible animals, and I believe has a very good chance to become new.
ADURO BIOTECH/CHRISTIAN LEE/GENOMICS INSTITUTE OF THE NOVARTIS RESEARCH FOUNDATION
Scientists have identified a small molecule drug that might prevent and treat Covid-19 by targeting a cellular gene that bypasses SARS-CoV-2 blockade of the innate immune response. The drug, diABZI, is currently undergoing phase II clinical trials as a potential cancer treatment.
So far, the search for Covid-19 drugs has yet to yield a product comparable in efficacy and utility to the mRNA vaccines created by Moderna and Pfizer. Remdesivir and monoclonal antibodies, the two FDA-approved treatments we currently have on hand, may be moderately effective, but they can only be administered intravenously in hospital settings.
Early on in the pandemic, experts thought interferon-based drugs would make a potent Covid-19 treatment. Interferon is a signaling protein the body relies on to activate critical immune sensing pathways, and studies showed that SARS-CoV-2 either actively inhibited or evaded it. If a virus goes to great lengths to avoid triggering interferon, it makes logical sense that loading up on interferon would succeed as a countermeasure. But clinical trials testing interferon drugs fell short of expectations, yielding mixed results.
Interferon may have been an obvious choice, but obvious in theory doesn’t always translate to success in practice. The problem isn’t necessarily that interferons don’t work. Using them so directly might just be too blunt an approach. New research, published in the journal Science Immunology in May 2021, suggests that using a small molecule drug to preemptively trigger the innate immune response downstream from interferons—specifically, the STING pathway—might do the trick instead.
Immunity typically transpires in two waves. The first line of defense the virus encounters when it enters the body is the innate immune response, followed by an adaptive immune response that mobilizies T cells and B cells to recognize and attack specific pathogens. Innate immunity is necessary to trigger adaptive response.
Innate immunity involves proteins known as pattern-recognition receptors that, over the course of evolution, have been hard-wired to identify pathogen-associated molecular patterns, or PAMPs. Some of the better known PAMPs include bacterial lipopolysaccharides, acids, bacterial DNA, and both single- and double-stranded RNA.
A Science Immunology study led by microbiologist Sara Cherry and her colleagues at the University of Pennsylvania is the latest to show that the virus can evade these receptors adroitly enough to delay innate immunity for a significant period of time. To document this delay, the team infected respiratory cells with SARS-CoV-2 and monitored them for antiviral activity. A full 24 hours later, the cells exhibited very few transcriptional changes. It took 24 hours more—48 hours total—for innate immunity to kick into gear and begin expressing interferon-stimulated genes.
The suppression of gene expression is evidence of immune evasion, but not necessarily antagonism—a recurring theme throughout this research paper. The 24-hour timeline may be hypothetical, but the evolutionary advantage these mechanisms give the virus is not. So long as SARS-CoV-2 eludes detection, it can replicate unimpeded across the ACE2 receptors of bodily tissues and exit the body via nasopharyngeal and gastrointestinal passages, dispersing into the air and entering new hosts.
Across several more laboratory experiments, Cherry and her research team observed SARS-CoV-2 evading immune sensing pathways, interferon activation, and detection by pattern-recognition receptors. If a drug could stimulate these pathways independently, they reasoned, it might bypass viral suppression of the initial immune response. A second Science Immunology study, released at the same time as Cherry’s but conducted by a team from the University of Massachusetts, had the same hypothesis.
The target selected for both studies was the Stimulator of Interferon Genes, otherwise known as STING. Interferons induce STING, which in turn activates a cascade of downstream factors, including STAT6 and IRF3, through the coding gene TBK1. Activation of the STING pathway leads to activation of both the innate and adaptive immune response, suppressing viral replication before it can overwhelm our bodily systems.
Critically, the STING pathway appears to bypass the many blockades SARS-CoV-2 mounts against the innate immune response. Spurred by this observation, Cherry and her team screened 75 potential STING agonists for their ability to block SARS-CoV-2. Of all the candidates, nine suppressed infection more than tenfold. Two cyclic dinucleotides showed particular promise but, as negatively charged molecules, they crossed the cell membrane at too inefficient a rate to warrant further examination.
Instead, Cherry and her team chose diamidobenzimidazole, or diABZI, a small molecule drug that mimics cyclic dinucleotides in most respects—except it readily crosses the cell membrane. It is currently in trials for cancer treatment. The University of Massachusetts researchers chose diABZI-4, a new compound of the same drug. Both studies found that diABZI potently inhibited infection in both cell cultures and mice models, inducing the expression of hundreds of antiviral genes the virus would otherwise delay.
Cherry and her team treated respiratory cells with diABZI and observed that it induced expression of more than 400 genes, nearly 40 percent of which link back to interferon signaling pathways. Compared to cells treated with interferon beta, upregulation of interferon-stimulated genes in diABZI-treated cells was more transient—meaning the risk of overstimulation is much less. Despite differences in duration, diABZI still suppressed the virus at a potency comparable to type I interferon treatment, blocking SARS-CoV-2 replication about 1000-fold in air-liquid interface cultures.
In mice models, the results were even more promising. A single dose, delivered intranasally six hours prior to infection, helped prevent early weight loss and enable their full recovery. Rather than an oral or intravenous point of entry, the researchers focused on the respiratory tract because it is typically the first site of infection and, once the immune system is activated, the first line of defense. In subsequent dose-response experiments, even the lowest dose of diABZI resulted in about 1000-fold inhibition. The potency of the immune reaction triggered by diABZI, the researchers also found, was as robust against the B.1.351 (South Africa) strain as the original (Wuhan) strain. This suggests the treatment might be effective at deterring respiratory viruses more broadly.
The Science Immunology studies mostly focused on diABZI as a potential prophylactic—a drug administered prior to infection, with the aim of prevention. But given to the mice therapeutically, with the aim of mitigating the severity of symptoms post-infection, diABZI still proved effective at reducing viral loads. Furthermore, unlike monoclonal antibody drugs, use of a chemical-based drug like diABZI isn’t affected or restricted by antibody resistance.
These are just the kinds of drug treatments we need to deter a virus as wily and elusive as SARS-CoV-2—powerful, broadly protective, and capable of disarming the virus with molecular precision, from multiple directions.
Originally published on Forbes (June 3, 2021)
Read Dr. Haseltine's latest piece with
![]()
