A Neanderthal Gene That Protects Us From Covid-19 (Part 14)
(Posted on Wednesday, October 6, 2021)
This is the fourteenth article in a series called “How SARS-CoV-2 Delays, Evades, and Suppresses the Immune System.” Read parts 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, and 13.

OAS1 as St. Michael, slaying the dragon SARS-CoV-2 via multiple points of attack. “Archangel Michael Slaying the Dragon” Master of Saint Verdiana (Italian, active ca. 1380-ca. 1420)
THE WALTERS ART MUSEUM HTTPS://ART.THEWALTERS.ORG/DETAIL/37741/ARCHANGEL-MICHAEL-SLAYING-THE-DRAGON/
Science continues to unveil the secrets of Covid-19 one paper at a time, decoding how the virus works and what the body does to protect itself in response. A new study offers a deep glimpse into this perennial battle between humans and infectious diseases by way of a gene that appears to have the power to knock out SARS-CoV-2.
One of the fundamental mechanisms the body uses to destroy incoming threats is attacking the genome. Since the SARS-CoV-2 genome consists of RNA, our cells can digest it. But that same ability isn’t tightly controlled, it can become deleterious to the host, digesting cellular, rather than viral, RNA.
What unleashes the destructive force that can potentially annihilate the cell? In the case of SARS-CoV-2, and for many other viruses as well, it is ribonuclease L, which normally exists as a single entity but becomes active when it receives a specific signal and forms a dimer that can digest RNA both viral and cellular.
What triggers RNAse L? The short answer is interferon. Whenever a virus invades a human cell, a series of pathways comparable in complexity to a Rube Goldberg apparatus is activated to induce interferon production (see Figure 1). Interferon in turn induces hundreds of interferon-stimulated genes. Viruses like SARS-CoV-2 do their best to prevent this cascade from happening, but their attempts at blockade can only go so far or last so long.
In a recent publication in Science, Wickenhagen et al. show that one interferon-stimulated gene, 2’-5’ oligoadenylate synthetase (OAS1), controls SARS-CoV-2 infections. The OAS1 protein exists in two forms: a longer 46 thousand dalton (46K) protein and a shorter 42K version. The ability to make either the long 46K or short 42K form of the OAS1 protein is genetically determined. The longer 46k form carries a signal that allows it to be modified by the addition of a lipid, a process called prenylation. The addition of the lipid allows OAS1 to associate with cellular membranes. It is the longer 46K prenylated form of OAS1, not the shorter 42K unprenylated version, that provides the signal that activates ribonuclease L upon SARS-CoV-2 infection.
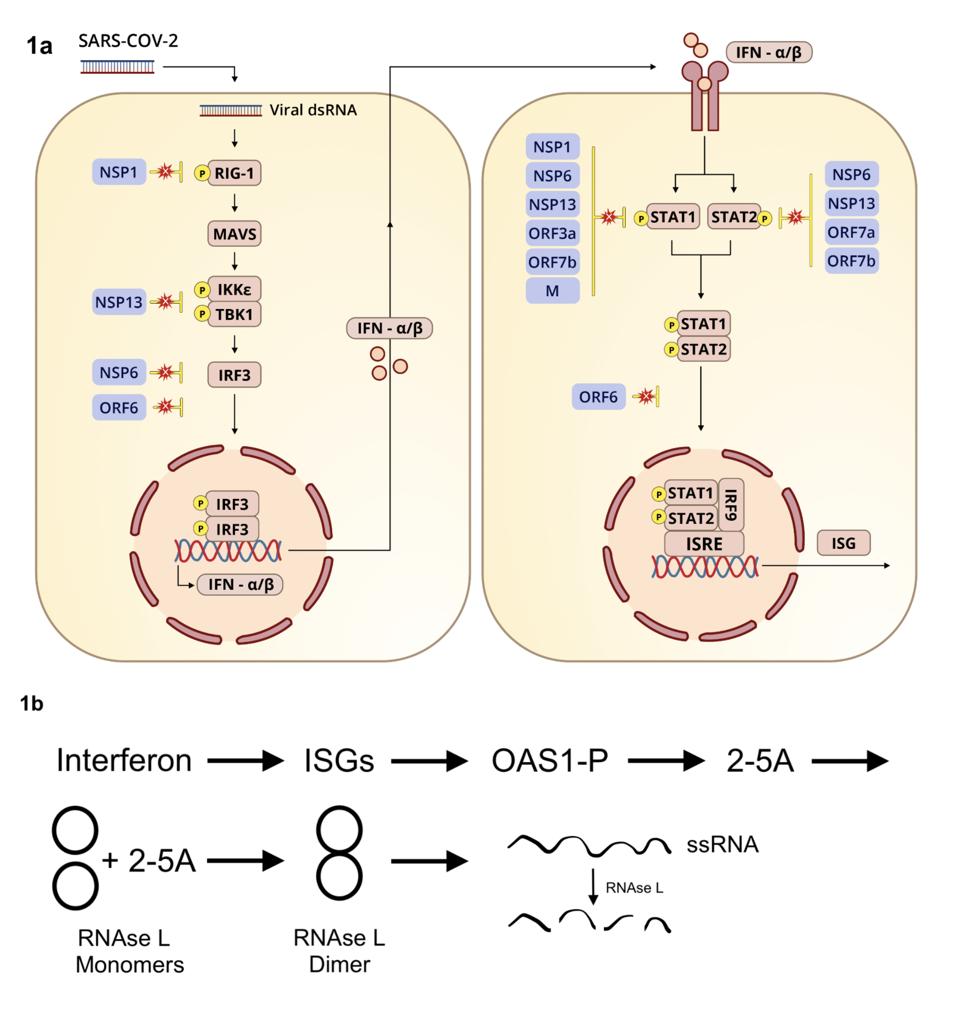
Figure 1. (a) Diagram illustrating induction of interferon-stimulated genes. (b) Interferon stimulation of RNA degradation via OAS1 activation.
ACCESS HEALTH INTERNATIONAL
In another tour de force, Wickenhagen et al. decipher the precise signal that triggers OAS1 activity. Previous studies show that OAS1 is activated when it detects double-stranded RNA. How then do the single stands of viral and messenger RNAs active OAS1? The answer is unexpected. OAS1 binds to two short segments of viral RNA near the 5’ end of the viral RNA called stem-loops 1 and 2. By pairing with itself, this region of the SARS-CoV-2 genome forms the requisite double-stranded stem-loop structures (Figure 2). The authors reveal the intimate interactions between the stem-loops at the beginning of the viral genome and viral messenger RNAs. Once OAS1 binds to the stem-loops 1 and 2, it produces a small molecule called 2’-5’-oligoadenylate (2-5A). It is 2-5A that activates RNAse L. In the presence of 2-5A, inactive monomers of RNAse L—held in reserve in case of viral attack—dimerize to form the active enzyme that shreds the viral RNA.
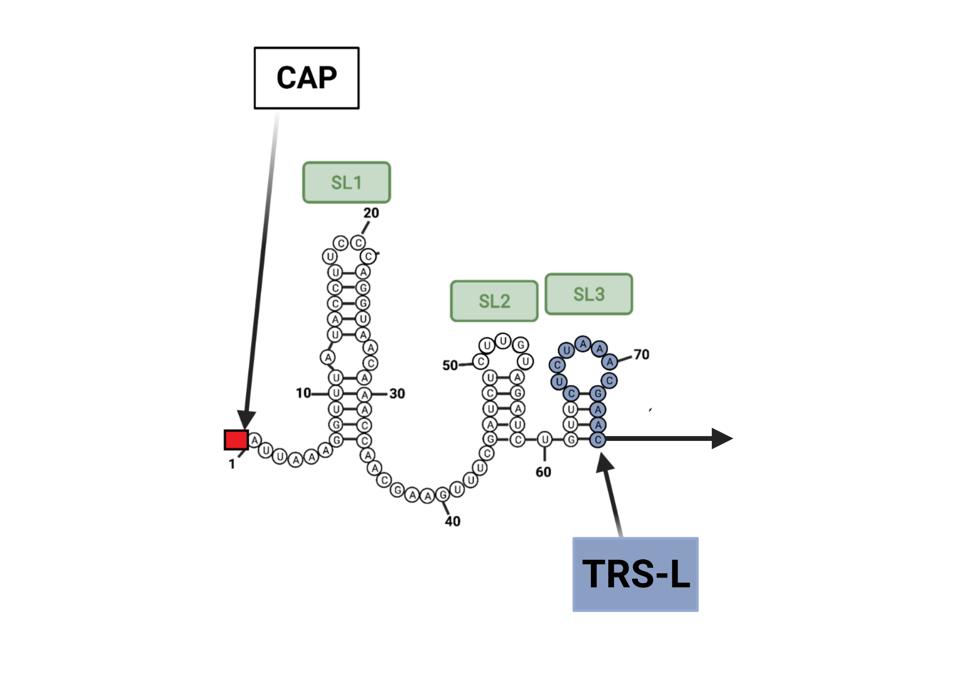
Figure 2. OAS1 binds to two short segments of viral RNA near the 5’ end of the viral RNA called stem-loops 1 and 2. By pairing with itself, this region of the SARS-CoV-2 genome forms the requisite double-stranded stem-loop structures. TRS-L is the
ACCESS HEALTH INTERNATIONAL
Prenylation is key to OAS1 recognition of viral RNA. Virus replication occurs in a special compartment walled off from the rest of the cell by a double membrane vesicle. The researchers show that prenylated 46K OAS1 binds to the double-membrane vesicle whereas the unprenylated 42K form does not. In other words, the longer form of OAS1 penetrates one of the virus’s primary defenses, the shield of protection afforded by the highly structured replication compartment. Wickenhagen et al. report that the double membrane replication compartment is fragmented by the active OAS1-RNAse L duo. The authors find that the 46K version of OAS1 inhibits many other pathogenic viruses that like SARS-CoV-2 replicate within their own double-membrane vesicle concealment, including encephalomyocarditis virus and some other coronaviruses.
We have known for some time that ribonuclease L is an enemy of SARS-CoV-2. This series describes the many ways by which the SARS-CoV-2 inhibits interferon and interferon stimulated genes (Figure 1). Wickenhagen et al. of this study have advanced our understanding of one of the primary reasons why SARS-CoV-2 must deactivate interferon and interferon induced genes to survive.
Why is this important? Wickenhagen et al. confirm that those who inherit the 42K version of OAS1 are much more likely to end up severely ill, in the ICU, or dying from Covid. Sequencing samples from almost 500 hospitalized Covid-19 patients, the authors of the study found that the median transcript abundance of 46K OAS1 was lower in patients with severe Covid-19 by more than 100-fold. The long 46K version of OAS1 is what we might call a survival gene (more technically a survival allele). It should be possible to test incoming patients for this inherited trait to predict disease outcomes.
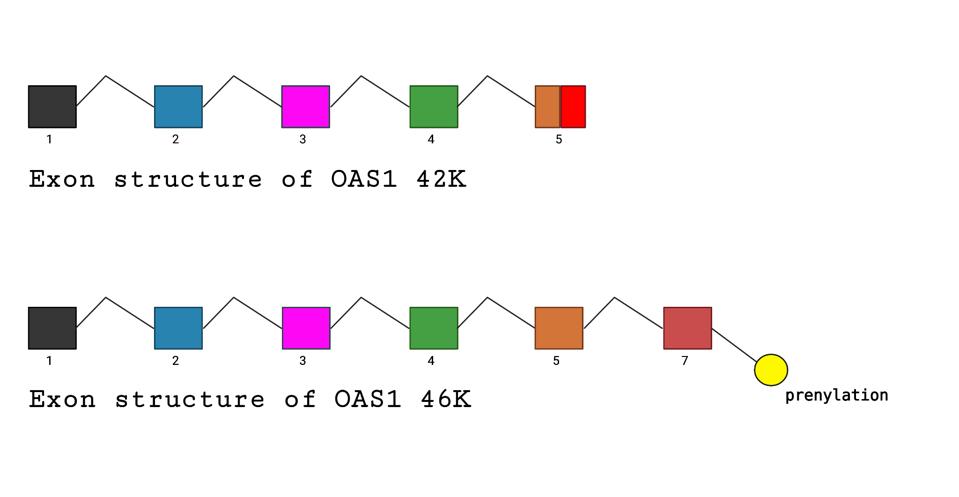
Figure 3. Comparative diagrams of exon structures of 42K and 46K OAS1. The difference in the alleles that specify the short or longer, prenylated forms of OAS1 are determined by a single nucleotide difference in the splice acceptor site of exon 7.
ACCESS HEALTH INTERNATIONAL
Population studies show that this Covid-19 survival allele is not evenly distributed in populations throughout the world. At the low end of the spectrum is a cohort from Lima, Peru at 11 percent; at the high end, another from Esan, Nigeria at 70 percent. Most people in the United States lack the protective variant of OAS1 (Figure 4). It might be worth knowing your person of the OAS1 gene to understand at least one component of your risk from Covid-19.
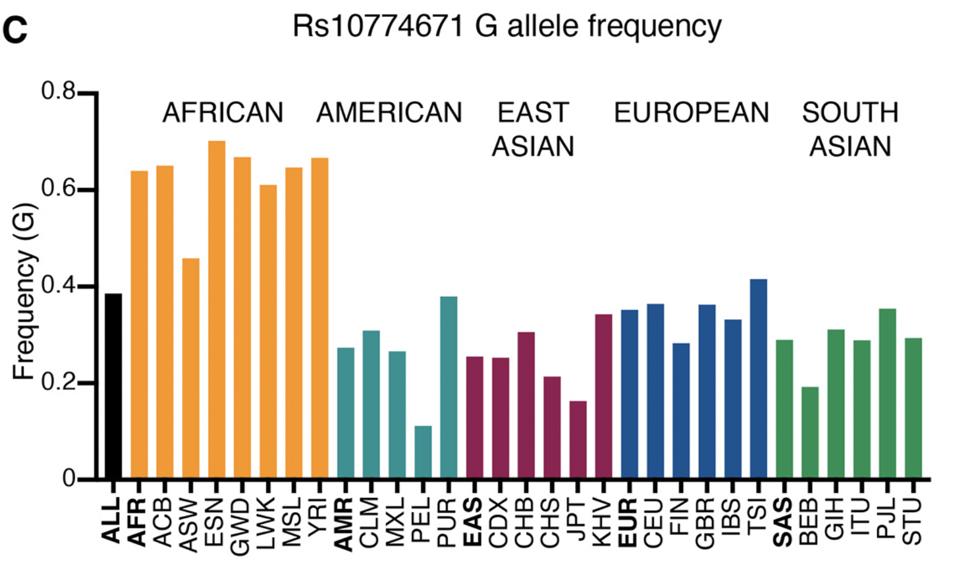
Figure 4. Frequency of alleles with G at Rs10774671 in different human populations (1000 genomes project). The population names are expanded in the materials and methods.
SOURCE: HTTPS://WWW.SCIENCE.ORG/DOI/10.1126/SCIENCE.ABJ3624
Wickenhagen et al. also confirm an early observation that the protective form of OAS1 is inherited from our Neanderthal ancestors. It is now well established that up to two to five percent of our genes survive from interbreeding with our Neanderthal predecessors. The 46K variants is one of those traits. It seems likely that prevalence in the population reflects successful defense against past and present encounters with lethal viruses.
Note that two viruses, MERS-CoV and SARS-CoV, are resistant to RNA cell defense OAS1. We know how MERS-CoV and at least one of the cold-causing coronaviruses evade RNA cell destruction. They encode a protein, NS4b, that cleaves 2-5A, limiting RNAse L activation. At present it is unknown how SARS-CoV-1 evades RNAse L as it has no protein equivalent to the other coronavirus NS4b. Nonetheless evasion of this pathway appears to increase lethality. SARS and MERS kill approximately 10 percent and 30 percent of those infected, respectively. We can only hope SARS-CoV-2 never acquires the accessory the ability to inactivate 2-5A either by evolution or recombination from bat or cold-causing human coronaviruses.
The authors also describe several other interferon stimulated genes that inhibit SARS-CoV-2 replication. A deep exploration of the role of these genes and others yet to be discovered is needed.
A final note. Drug candidates exist that mimic 2-5A. Such drugs may help Covid-19 patients who cannot fight the virus on their own.
Originally published on Forbes (October 5, 2021)
Read Dr. Haseltine's latest piece with
![]()
