Flying Under The Radar: How SARS-CoV-2 ORF7a Contributes To Immune Evasion And Inflammation
(Posted on Monday, June 20, 2022)
This article is an extension of our series on immune suppression by SARS-CoV-2. The series has since been published as a book, Natural Immunity and Covid-19: What it is and How it Can Save Your Life. It is also available to read on my website. Here, we discuss new data regarding the ORF7a viral protein. Click for part 1.
The success of SARS-CoV-2 as a virus hinges in large part on its ability to suppress and evade our immune system. Usually, our immune system encounters an invading microbe and immediately springs into action. But SARS-CoV-2 is a master of flying underneath the radar, staying hidden from our innate immune response. It also evolves rapidly to evade our adaptive immune response.
Interferons —a group of signaling proteins produced by cells in response to microbial threat— are key to both the innate and adaptive immune response. In a previous article, we outlined how ORF7a inhibits the induction of interferon-stimulated genes by preventing the phosphorylation of STAT2. This obstructs our interferon response, making it more difficult for our immune system to mount a successful counteroffensive. More recently, we described the discovery that ORF7a also blocks the antiviral function of a host protein called SERINC5, which would otherwise help prevent viral entry into cells.
Covid-19 Severity: Monocytes and Inflammation
Most people who contract Covid-19 will suffer only mild, if any, symptoms. But there is a subset of patients who go on to develop severe disease — with upwards of half a billion confirmed cases, this subset quickly becomes very large. Severe cases are generally characterized by an overactive inflammatory response associated with elevated cytokine levels, a drop in the number of white blood cells, and infiltration of macrophages and monocytes into different tissues. Monocytes, in particular, appear to hold an important role in the hyperinflammation seen in severe Covid-19 cases.
Monocytes are large white blood cells that circulate the body through the bloodstream, keeping an eye out for any microbial threats. They recognize microbes via pattern recognition receptors (PRRs) that line their surface. These receptors pick up on age-old molecular patterns typical of pathogens — be they viral, bacterial, fungal, or parasitic. Once they have spotted a pathogen, they migrate to the affected area and help stimulate the inflammatory response by producing cytokines. Monocytes can also differentiate into two other types of immune cells: phagocytes, which engulf and destroy microbes, and dendritic cells, which present T cells with antigens to help stimulate a more specific immune response. As such, suppression of monocytes can lead to all kinds of knock-on immune dysregulation.
Although aberrant inflammation and upregulation of monocytes had been associated with worse Covid-19 outcomes, the viral proteins responsible for this excessive inflammation remained largely unknown.
ORF7a: New Structural Insights
Certain proteins contain immunoglobulin-like (Ig-like) molecular structures. These play a critical role in modulating interactions in the immune system. Usually these are host proteins, such antibodies. Certain viruses have developed proteins with similar structures to help them hijack the host immune system.
Zhou et al. scanned SARS-CoV-2 proteins for Ig-like structures and noticed that ORF7a contains an Ig-like ectodomain — the section of a protein that extends out from the surface and initiates contact with other proteins and cells. The ORF7a ectodomain is made up of seven beta strands (β-strands) that form two connected beta sheets (β-sheets) (Figure 1). The resultant structure looks like a hand, palm facing inwards. It is the “fingers” of this hand that end up reaching out and binding to other proteins.
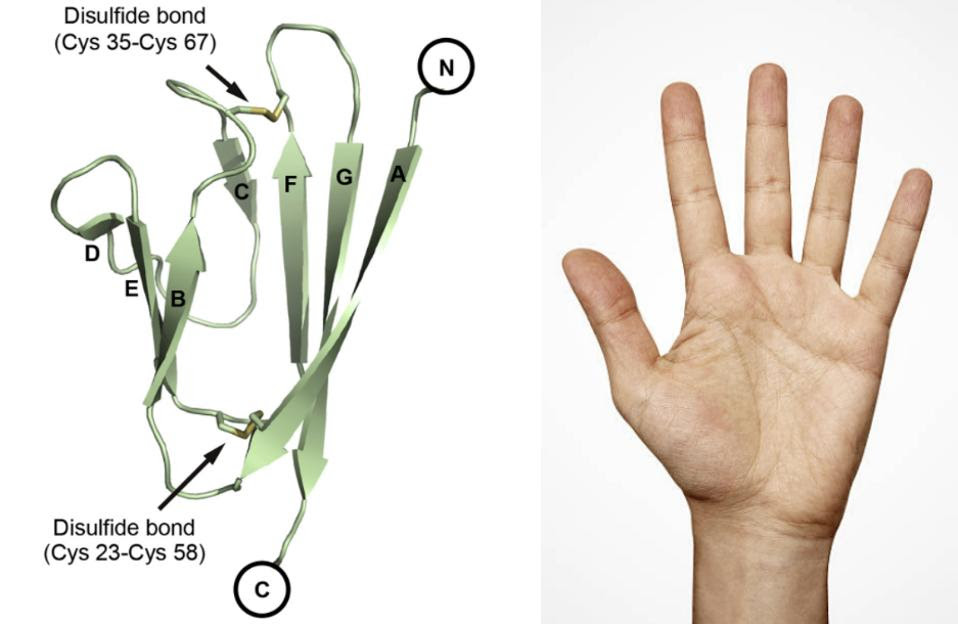
FIGURE 1. (LEFT) Ribbon representation of a SARS-CoV-2 ORF7a molecule. Disulfide bonds are … [+] FROM: (LEFT) ZHOU ET AL. 2021 (RIGHT) GETTY IMAGES
ORF7a contains an Ig-like ectodomain, but does it actually interact with host immune cells? To find out, the researchers exposed human peripheral blood mononuclear cells (PBMCs) —lymphocytes and monocytes— from healthy donors to SARS-CoV-2 ORF7a. They discovered that ORF7a binds to CD14+ monocytes with a high degree of efficiency. It also binds to lymphocytes, but much more weakly.
SARS-CoV-1, the virus responsible for the 2003 SARS outbreak, also has an ORF7a protein. The sequence similarity between the two is 87% and they share a very similar structure as well. Still, the ORF7a protein of SARS-CoV-1 binds only weakly to monocytes. The researchers used this difference in binding affinity to identify the precise structural features of SARS-CoV-2 ORF7a that allow it to bind so efficiently. They discovered that the two ORF7a proteins differ from one another in the distribution of the binding site residues. Compared to SARS-CoV-1 ORF7a, the binding residues of SARS-CoV-2 ORF7a were all located on the larger beta sheet (strands A, G, F, and C). None were located on the smaller, three-strand beta sheet (strands D, E, and B). This implies the key binding residues are located on the larger beta sheet (Figure 2).
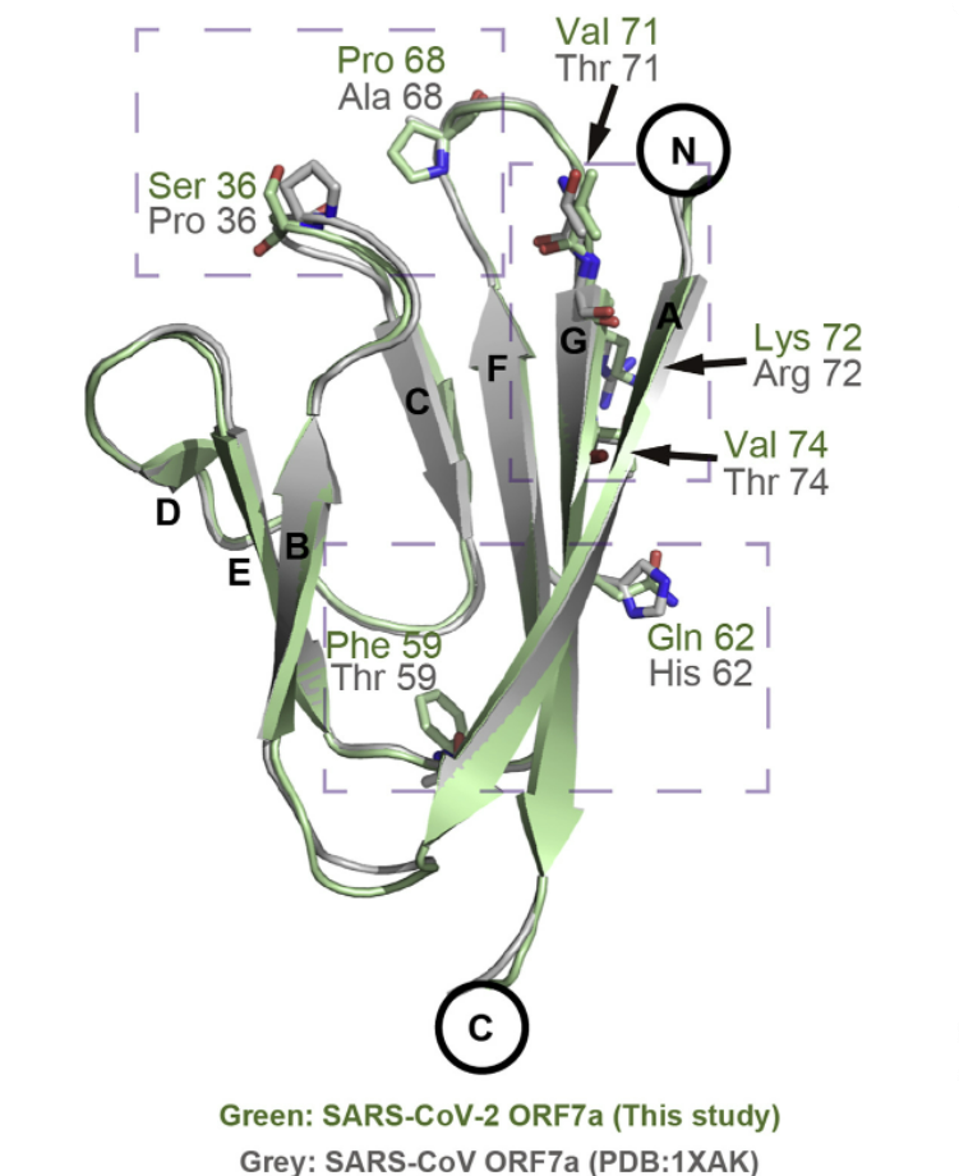
FIGURE 2. Structural alignment between SARS-CoV-2 ORF7a (this study) and SARS-CoV ORF7a (PDB: 1XAK). … [+] FROM: ZHOU ET AL. 2021
ORF7a Suppresses Monocytes
Having established that ORF7a can bind to monocytes and lymphocytes, the researchers looked into the effects of this interaction. They co-incubated SARS-CoV-2 ORF7a with human monocytes and lymphocytes for a 24 hour period. Zhou et al. then measured the expression levels of human leukocyte antigen (HLA) surface molecules on the immune cells.
The HLA system is used to present antigens to other cells, and is a critical component of the adaptive immune response. It can be broken down into two classes. First, those proteins that help to move antigen snippets from the inside of a cell to the cell surface. This signals to the immune system that a cell has been infected and needs to be destroyed, thereby slowing the spread of the pathogen. This class is made up of HLA-A, HLA-B, and HLA-C. The second class is composed of HLAs that present antigens found outside of the cell to T-helper cells. The T-helper cells then go on to stimulate the production of B-cells, which secrete antibodies specific to the microbial threat at hand. This class includes HLA-DR, HLA-DP, and HLA-DQ.
The researchers noticed a significant decrease in the expression of the second kind of HLAs on the surface of CD14+ monocytes — roughly 30% lower than normal. There was no difference in the expression of HLA-A/B/C. The expression of HLAs on the surface of lymphocytes remained unchanged.
So ORF7a decreases the amount of HLA receptors on the surface of CD14+ monocytes, hindering their ability to signal for additional support from other immune cells. Zhou et al. conclude that this is likely an immune escape tactic on behalf of SARS-CoV-2 — essentially, buying it more time to spread unnoticed. The precise mechanism through which the ORF7a ectodomain modulates the antigen-presenting ability of CD14+ monocytes remains to be determined.
ORF7a Triggers Inflammation
As mentioned, hyperinflammation is a hallmark of Covid-19. In a previous article we discussed how SARS-CoV-2 can infect monocytes, leading to a form of cell death called pyroptosis and the sudden release of a mass of pro-inflammatory cytokines. Findings by Zhou et al. implicate ORF7a as an additional source of monocyte-mediated inflammation.
To study the effects of ORF7a on inflammation, the team of scientists tested blood samples for markers of inflammation. They discovered that co-incubation with ORF7a triggered a sharp increase in the production of proinflammatory cytokines. More specifically, they noticed that co-incubation led to the upregulation of those cytokines most closely associated with cytokine storms — IL-6, IL-1β, IL-8, and TNF-α. The spike in cytokine production implies that ORF7a immune modulation of monocytes may be a contributing factor to the hyperinflammation seen in severe Covid-19 cases.
Interestingly, the cytokine profiles differed depending on the donor. Some blood samples had a more intense cytokine response than others. This indicates there may be genetic differences governing ORF7a interaction and, by extension, disease outcome. Future research should aim to pinpoint the factors responsible for the variation in cytokine response.
Why Inflammation?
ORF8, another SARS-CoV-2 accessory protein, is also known to trigger inflammation. In this case, the process is very targeted, with ORF8 mimicking one of our immune system’s most powerful inflammatory response triggers, interleukin-17.
The question arises, is the inflammation triggered by SARS-CoV-2 infection simply an unfortunate side effect, or might it be to the benefit of viral replication? One observation that supports the hypothesis that inflammation may contribute to the success of the virus is that the receptor for viral entry, angiotensin converting enzyme 2 (ACE2), is induced as part of the inflammatory response in endothelial and other cells. A release of inflammatory substances may therefore be advantageous, and not entirely adventitious.
The ability of ORF8 and ORF7a to, in and of themselves, induce an inflammatory response favors the advantageous hypothesis.
Take-home Message
This work by Zhou et al. opens the door to a new target for drug development, ORF7a. It also adds to the growing list of reasons to shift our focus onto the entirety of the SARS-CoV-2 genome, not just the Spike protein. There are many accessory proteins that contribute to the virus’ ability to evade and suppress our immune system; the sooner we learn about their role in infection, the sooner we can begin creating effective anti-Covid drugs.
Read Dr. Haseltine's latest piece with
![]()
