JN.1: The Odd Man Out Among Omicron Sublineages
(Posted on Thursday, October 26, 2023)

This article was originally published on Forbes on 10/26/23.
A new Covid-19 variant demonstrates the rapid adaptability of the SARS-CoV-2 virus. This latest emerging SARS-CoV-2 variant, JN.1, was identified in Luxembourg on August 25, 2023, followed by England, Iceland, France, and the United States. In the GISAID SARS-CoV-2 database, there are 91 instances of JN.1 at the time of writing, suggesting that many hundreds, if not thousands have been infected with the variant as sequencing efforts have been largely halted.
What is most intriguing about JN.1 and why I believe there is cause for concern is the striking amount of differences between it and the leading viruses today: XBB.1.5 and HV.1. The XBB.1.5 variant is the target of the latest vaccine boosters in the United States. Most emerging variants are descendants of this virus. HV.1 is a relative newcomer and boasts a few differences from XBB.1.5 but is mainly similar to XBB.1.5.
To quantify the departure of JN.1 from these viruses, there are ten additional unique mutations in HV.1 compared to XBB.1.5, or a difference of about 12%. JN.1 contains 41 additional unique mutations compared to XBB.1.5, or 40%. Most of the changes in JN.1 are found in the spike protein, which likely correlates to increases in infectivity and immune evasion.
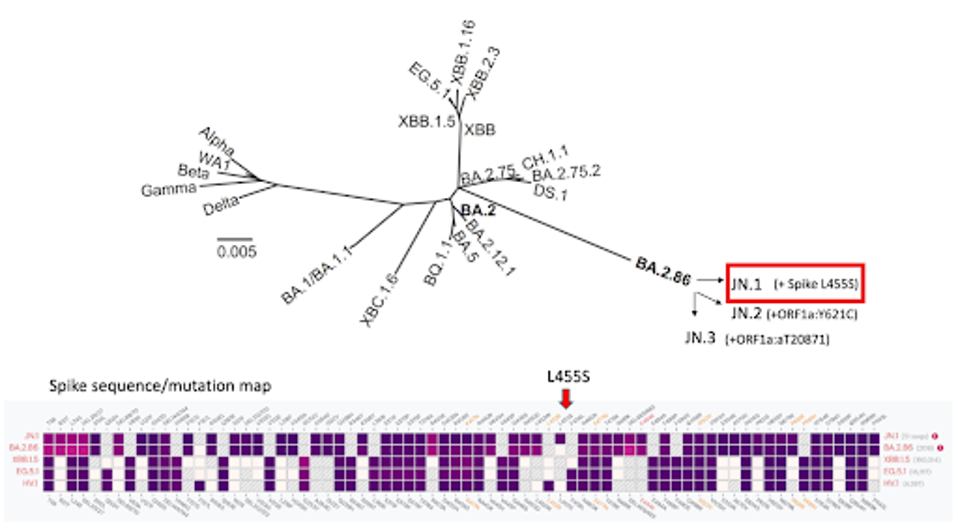
FIGURE 1: SARS-CoV-2 lineage map. Note the emergence of JN.1 and others from BA.2.86.
Many spike protein mutations are among those we have seen in previous variants of concern, such as E484K and P681R, which were first identified in the Alpha and Beta variants of SARS-CoV-2 in early 2021. Why those mutations fell out of circulation in the Omicron family of variants is unclear, but their reemergence in JN.1 is noteworthy.
What catches my attention about JN.1 is not the reemerging mutations from earlier variants but the novelty of select mutations in the spike protein. Several of these mutations have only been sequenced a few thousand times from a database of over 16 million samples throughout the pandemic. None, however, are unique to JN.1.
In the N-terminal domain, which plays a role in virus entry post-infection, mutations such as R21T, S50L, V127F, R158G, and others may improve viral entry efficiency, as well as immune escape from antibodies by implementing N-glycosylation sites.
In the receptor-binding domain, we observe a similar phenomenon. V445H, S450D, and L452W have all been sequenced less than two thousand times, among other rare mutations, all of which could work to improve ACE2 binding affinity or reduce antibody binding efficiency.
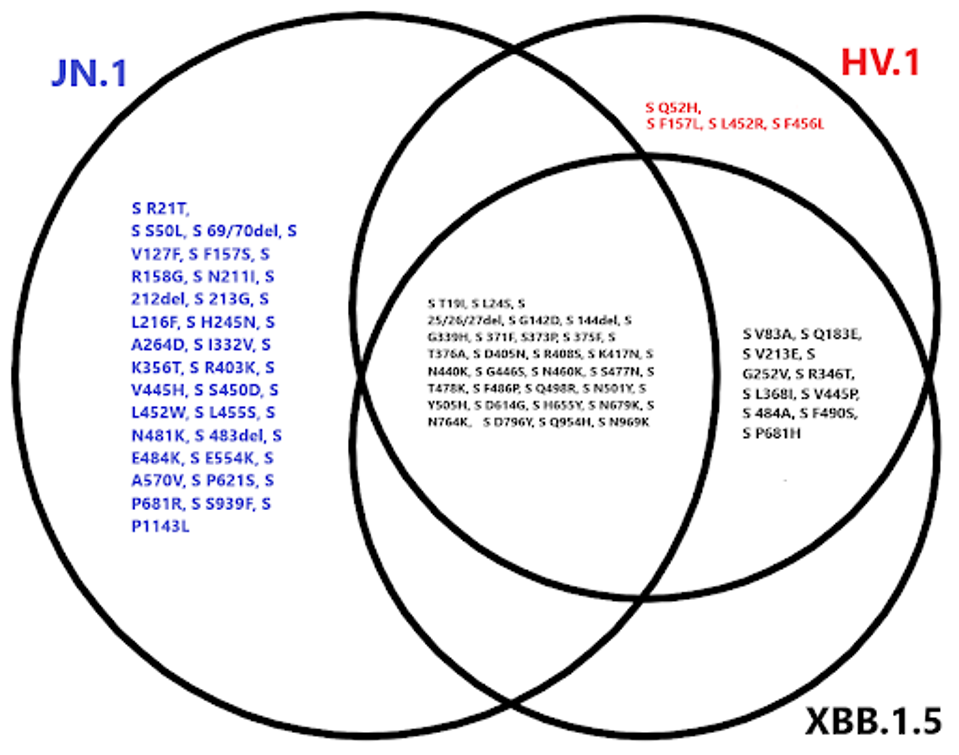
FIGURE 2: Spike protein mutational profile of JN.1 compared to XBB.1.5 and HV.1.
I want to draw your attention to mutations outside the spike region, which may significantly affect the virus’s pathogenicity and spread. Throughout the genome, there are a wide variety of mutations in the Orf1ab replication-transcription complex (NSP1-16), some in the structural proteins (E, M, and N), and a few in the accessory proteins (Orf3a-8). We bring attention to these because mutations in some of these proteins, particularly the N protein, can make a significant difference in virus replication.
Below is the entire catalog of mutations found throughout the virus.
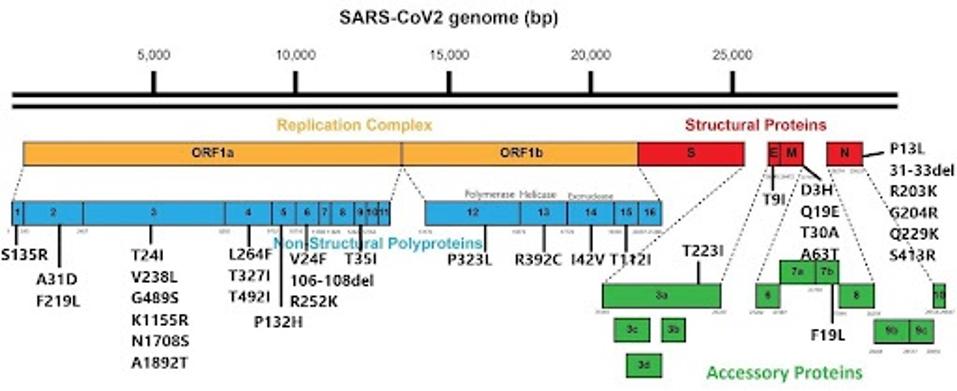
FIGURE 3: JN.1 nonspike mutations.

FIGURE 4: Nonspike protein mutational profile of JN.1 compared to XBB.1.5 and HV.1.
A nonspike protein that is heavily mutated in JN.1 is the NSP3 protein. There are six mutations in NSP3, namely T24I, V238L, G489S, K1155R, N1708S, and A1892T. NSP3 is one of the most active proteins in the virus, playing roles in viral RNA binding, polyprotein processing, and other functions. While the exact function of these mutations is unknown, they are likely to increase the efficiency of many of these mechanisms, creating a more functional and pathogenetic virus.
I also note the heavily mutated N protein. The mutations R203K and G204R have been mutated in most virus variants throughout the pandemic and likely improve viral replication rate. The other mutations in N may also work to improve viral replication.
While the Orf8 protein is truncated in the widespread XBB.1.5 variant, it is fully present in JN.1.
There are several explanations for the mutations within and particularly outside of the spike protein. The first is adaptation to more aggressive infectivity. The second is to escape from neutralizing antibodies. The third is adaptation to more efficient post-infection pathogenesis, including replication aided by mutations in the N protein. The fourth is immune evasion from T cell recognition. Dr. Gaurav Gaiha and colleagues identified early in the pandemic that some mutations reduced binding affinity to T cell epitopes. In JN.1, eight mutations added from the XBB profile are among those Gaiha identifies, suggesting they have been counterselected for T cell recognition.
I also note the presence of synonymous mutations or those that do not result in an amino acid change. There are likely several synonymous mutations littered throughout JN.1; however, collecting data on these mutations is much more complex than amino acid mutations.
While synonymous mutations do not impact the amino acid sequence of the virus, it does affect the tertiary structure of the virus’s RNA, which studies suggest can play a role in the adaptation of the virus to the human host environment. They may also play a role in the relative abundance of viral encoded protein and immune responses in the infected person.
For instance, a recent study on the SARS-CoV-2 N protein shows that the N-terminal domain of N recognizes and binds RNA sequences in the five prime untranslated regions of the virus. The N protein is involved in RNA transcription and genome packaging into virus particles, which play a crucial role in virus transmission. Thus, altering the structures of the five prime ends could impact the function and efficiency of N, impacting the overall viral function of SARS-CoV-2.
My colleagues and I will soon release a book on molecular biology concerning the SARS-CoV-2 virus (Patarca and Haseltine, to be published by Wolters Kluwer in 2024). In the book, we discuss the role of each protein in the pathogenesis of Covid-19. Over time, and with further advances in artificial intelligence technologies, we will better understand the impact of intricate mutations on protein function. The slightest change can have a massive impact on the protein structure and the tertiary structure of nucleotide sequences. Eventually, we will be able to predict the pathogenesis of a virus before it infects a host.
It remains to be seen whether JN.1 will cause a new wave of Covid cases akin to Alpha or Omicron in years past. With only 91 recorded instances of JN.1, this will likely be one of many variants throughout the pandemic to appear disconcerting but remain relatively minor. If GISAID data correlates to global cases, it seems JN.1 is beginning to slow down and plateau compared to earlier this month.
I cannot say what the pathology of JN.1 will be, as it is only observational and experimental at this point.
However, it is crucial to be aware of these threats before they become widespread, not after. As we enter the winter months, another wave of cases could likely occur, and JN.1 is an avenue for that to take place.
Read Dr. Haseltine's latest piece with
![]()
