"Naked" Self-Amplifying RNA Vaccine Shows Promise As A Booster, Inducing Strong Cytotoxic T-Cell Responses
(Posted on Monday, November 7, 2022)
This is the second of two articles on a temperature-sensitive self-replicating RNA vaccine; click here for part one. They form part of a broader series on next-generation mRNA vaccines. The first three installments can be read here, here, and here, respectively.
RNA vaccines have revolutionized traditional vaccine technologies. The current generation of Covid-19 mRNA vaccines rely on messenger RNA that encodes the viral spike protein encased in a lipid particle injected into the muscle. The results have been spectacular in producing powerful, but unfortunately, short-lived protection from infection. The search for even more effective, second-generation, mRNA vaccines is underway. Self-amplifying RNA vaccines, described in this series, offer one promising new approach. A recent preprint by Amano et al. describes one such approach using self-amplifying RNA with some surprising properties. The vaccine candidate uses natural messenger RNA with no modified bases. The mRNA is naked, unprotected by a lipid nanoparticle coat. Moreover, the RNA is injected directly into the skin instead of the muscle. Additionally, the self-amplifying vector is temperature sensitive, active only at the lower temperature of the skin, not at the higher temperature of internal organs and muscle.
In a previous article, I covered the how and why of the new vaccine — how it was designed, and the rationale behind it. Here, I take a look at some of the preliminary results, all derived from mouse models.
Induction of Robust CD8+ T Cell Immunity
The design of the initial vaccine candidate is illustrated in figure 1B. The vaccine is designed to express only a small part of the SARS-CoV-2 spike protein, the roughly 176 amino acid long receptor binding domain (RBD). By contrast, approved mRNA vaccines encode the entire 1273 amino acid long spike protein (Figure 1A). The receptor binding domain is the part of the spike protein that enables it to dock to our cells, initiating viral entry.
FIGURE 1. (A) Diagram of c-saRNA1-RBD vaccine design, including the mutation to NSP2 that confers temperature sensitivity and the antigen construct (CD5 signaling peptide fused to the N terminus of the receptor binding domain of the SARS-CoV-2 protein). (B) Representation of SARS-CoV-2 spike protein, including its various domains: NTD, N-terminal domain; RBD, receptor-binding domain; FP, fusion peptide; HR1, heptad repeat 1, HR2, heptad repeat 2; TM, transmembrane domain; CT, cytoplasmic domain. SOURCE: ACCESS Health International (Figure 1A adapted from “Structural and functional insights into the spike protein mutations of emerging SARS-CoV-2 variants” GUPTA ET AL. 2021)

The researchers administered the vaccine to mice intradermally, just underneath the surface layer of the skin, using a microneedle injector commonly used for cosmetic purposes. Fourteen days later, they drew blood samples and analyzed them for immune markers. They then administered a second dose two weeks later and again waited fourteen days before drawing blood samples for analysis.
Amano et al. discovered indicators that a strong, selective T-cell response had taken place (Figure 2). In particular, they noted a sharp increase in RBD-specific CD8+ cytotoxic T cells, also known as killer T cells, which bind to infected cells and break apart their cell membrane. This destroys the infected cells, stopping viral replication and curbing the spread of infection. The researchers also witnessed a robust type 1 CD4+ helper T-cell response. Type 1 helper T cells regulate immunity by activating macrophages, which devour foreign pathogens, and by activating killer T cells. This is in contrast to type 2 helper T cells, which instead regulate the immune response by prompting B cells to differentiate into antibody-producing plasma B cells. Markers for type 2 helper T cells suggested only weak activation.
FIGURE 2. (A) Experiment design. (B) Cellular immune responses two weeks after the first dose of the temperature-sensitive self-amplifying RNA vaccine or placebo vaccine (PBO). IFN-y acts as a marker for CD8+ and type 1 CD4+ T cells; IL-4 acts as a marker for CD4+ T cells. (C) Cellular immune responses two weeks after the second dose of the temperature-sensitive self-amplifying RNA vaccine or placebo vaccine (PBO). FROM: “Controllable self-replicating RNA vaccine delivered intradermally elicits predominantly cellular immunity” AMANO ET AL. 2022
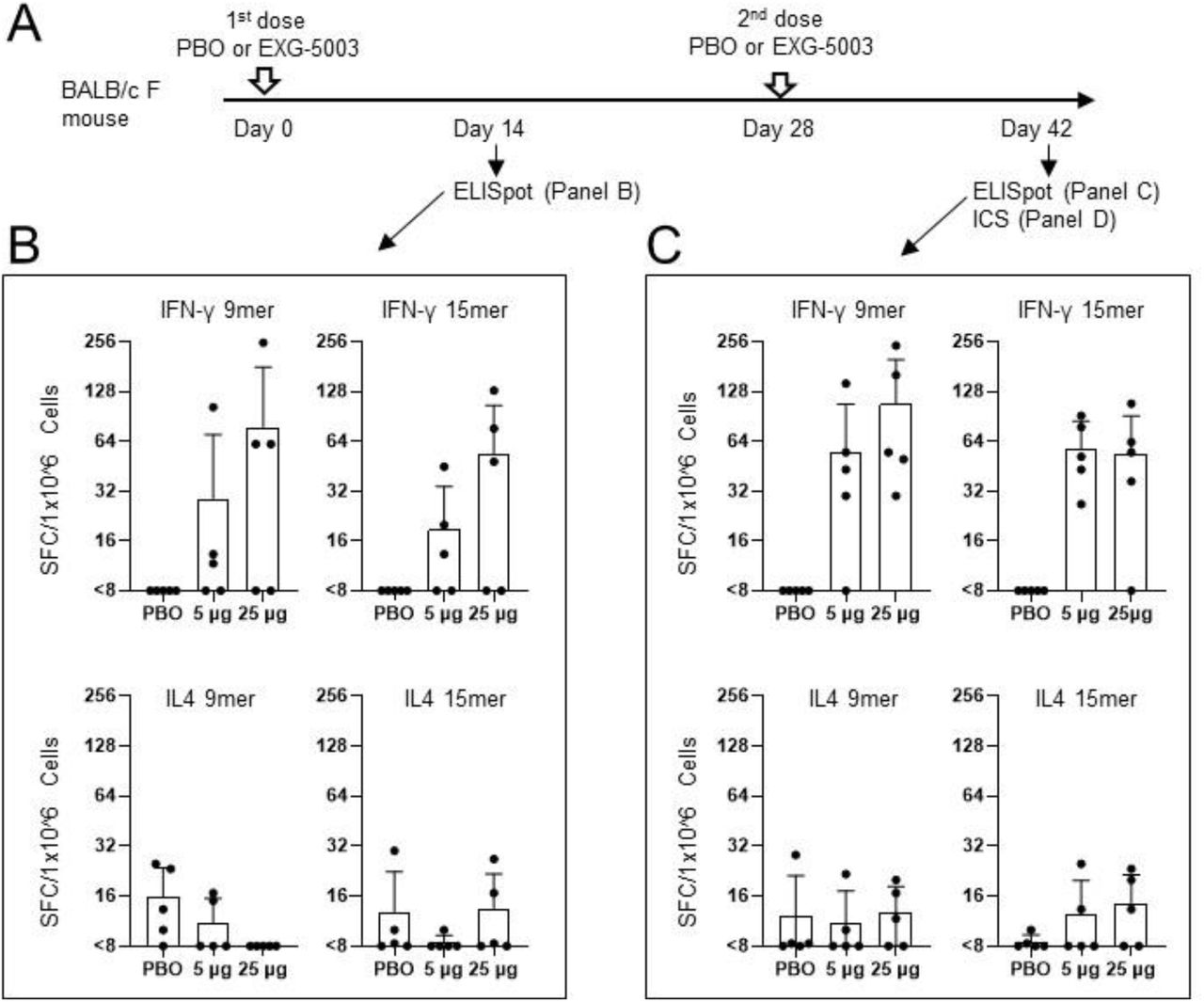
As expected, the weak type 2 helper T-cell response coincided with a lack of humoral immunity; the researchers did not find any RBD-specific antibodies following a two-dose regimen of their temperature-sensitive self-amplifying RNA vaccine.
Naked RNA for Prime/Boost Vaccinations
Many viral vaccines are spread across two or more doses. The first of these is referred to as a prime, and the follow-up dose is called a booster. Some vaccines may be better suited as a prime than a booster, or vice versa. Amano et al. tested their temperature-sensitive self-amplifying RNA vaccine in both capacities.
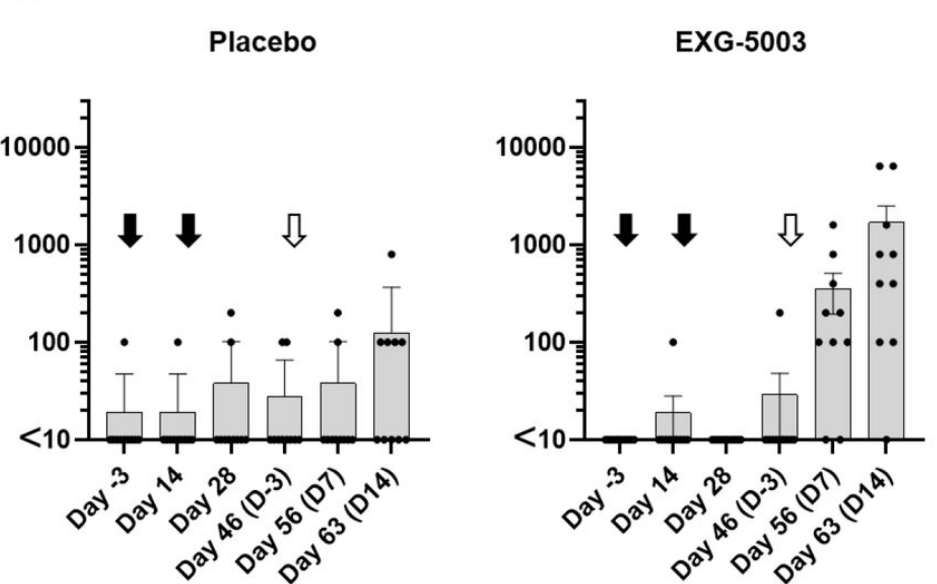
To see how it did as a prime, they first administered two doses of the vaccine fourteen days apart, drawing blood for analysis after each injection. They then waited three weeks before administering a booster dose of recombinant RBD protein. This was compared to a control group, which was given a placebo vaccine as a prime but the same RBD booster.
The mice receiving the temperature-sensitive vaccine displayed a rapid spike in RBD-specific antibodies following the booster dose (Figure 3). In the placebo group, the change in antibody titers was minimal. This suggests that, although the temperature-sensitive vaccine on its own fails to stimulate antibody production, it does manage to prime a secondary antibody response.
Interestingly, this held true even in mice exposed to a booster shot of the receptor binding domain of the Delta variant instead of the receptor binding domain of wild type SARS-CoV-2. Despite the structural differences between the two spike proteins, the vaccine still managed to prime the humoral response, with a noticeable increase in Delta-specific antibodies post-booster.
FIGURE 3. Antibody titers following a placebo vaccine/RBD boost regimen compared to antibody titers following a temperature-sensitive self-amplifying RNA vaccine (EXG-5003)/RBD boost regimen. Black arrows indicate the first two doses of vaccine (Day 0 and Day 14). White arrow indicates the administration of the booster dose. FROM: AMANO ET AL. 2022
Having shown their vaccine can act as a prime, Amano et al. next tested to see if it could be used as a booster shot to raise T-cell responses and stimulate a secondary antibody response.
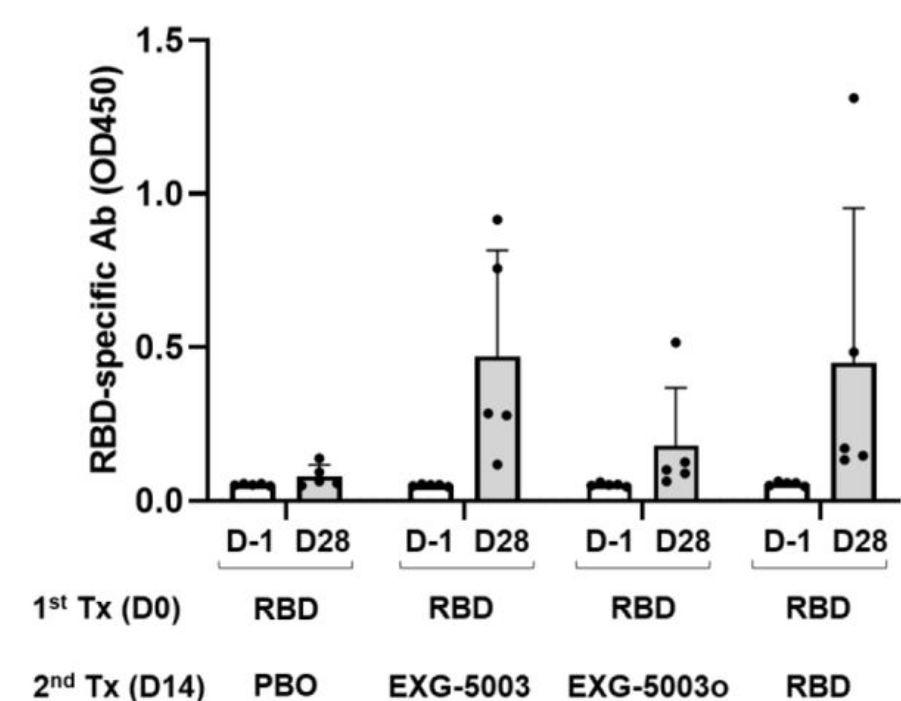
They injected mice intradermally with wild type SARS-CoV-2 RBD protein plus adjuvant as the prime vaccine. After two weeks, the mice were given a booster shot of either a placebo dose, a second dose of the RBD subunit vaccine plus adjuvant, or a dose of the temperature-sensitive self-amplifying RNA vaccine encoding the RBD. They also developed and tested an omicron-specific booster (EXG-5003o) by switching out the wildtype RBD of their self-amplifying RNA platform and replacing it with omicron RBD instead.
Mice that received the placebo booster did not manage to mount a cellular immune response. Those that received a second dose of the RBD subunit vaccine as a booster did mount a cellular immune response, but only weakly. As expected, the temperature-sensitive vaccine managed to induce robust T-cell responses. The omicron-specific booster induced the strongest T-cell responses.
The researchers also measured and compared antibody titers two weeks after the booster had been administered (Figure 4). Here, the temperature-sensitive booster managed to induce a humoral response on par with that of the adjuvanted RBD protein booster. The omicron-specific booster did not fare so well, likely because of the structural differences between the wild type and omicron spike proteins.
FIGURE 4. The levels of serum antibodies against the RBD protein of the SARS-CoV-2 (original strain), measured by an ELISA assay. The levels of antibodies are represented by the OD450 measurement. The average and standard deviation (error bars) of five mice (n=5) are shown for each group. The data of Day −1 (before the 1st treatment) and the data of Day 28 (after the 2nd treatment) are shown for each group. FROM: AMANO ET AL. 2022
Although the vaccine may not be best suited as a stand-alone vaccine it holds promise as a booster, successfully inducing both humoral and cellular immunity.
An Improved Vaccine
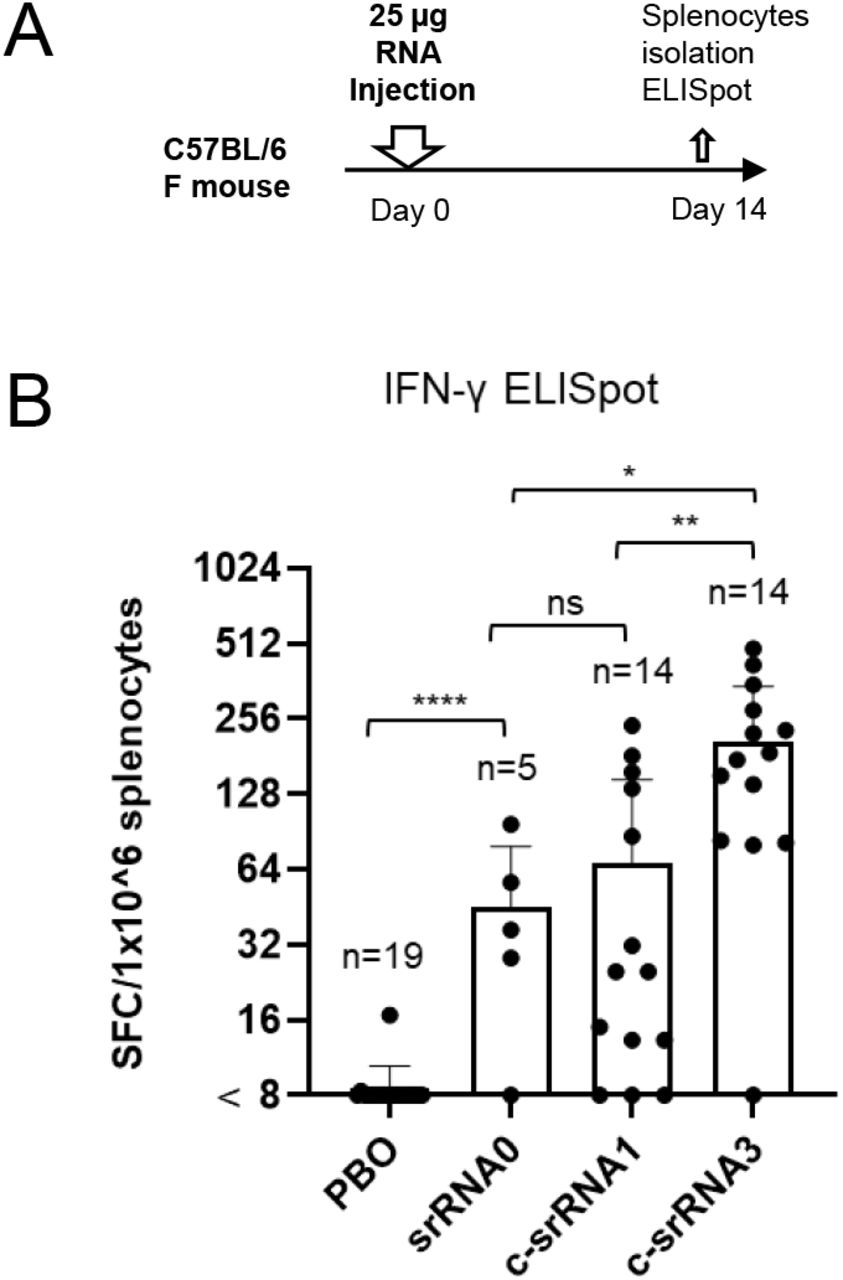
Amano et al. created a second version of their temperature-sensitive self-amplifying RNA platform in which they switched to a different viral vector. The original alphavirus vector they used contained a common mutation (A551D) as well as a truncated three prime untranslated region. For their new vector, they used an unmutated wild type strain of the virus, including a full-length three-prime untranslated region. The strain they settled on is especially adept at avoiding our innate immune response, giving the virus more time to spread the target antigen before being discovered.
The new vector design significantly boosted T-cell responses. Compared to the first version of their vaccine, the updated vector yielded a three-fold increase CD8+ killer T cells (Figure 5). And crucially, compared to a conventional, non-temperature-sensitive self-amplifying RNA vaccine, the researchers witnessed a six-fold increase in T-cell responses.
FIGURE 5. (A) Schematic of the experiment design (B) Graphical representation of T-cell responses post vaccination. From left to right: PBO, placebo; saRNA0, conventional self-amplifying RNA vaccine; c-saRNA1, initial iteration of the temperature-sensitive self-amplifying RNA vaccine; c-saRNA3, second iteration of the vaccine with improved viral vector (Trinidad strain of Venezuelan equine encephalitis virus). AMANO ET AL. 2022
Expanding Breadth: Testing a Nucleocapsid-based Vaccine
Despite the many uncertainties of the ongoing Covid-19 pandemic, one thing is clear: SARS-CoV-2 has the capacity to evolve and adapt. Wave after wave of infections driven by new variants confirms this. A key goal has been to develop vaccines that confer broad immunity that holds up to these continuous mutations. So called “universal” SARS-CoV-2 vaccines.
Amano et al. adapted their temperature-sensitive self-amplifying RNA vaccine platform to fit this need. Instead of targeting the receptor binding domain, which is part of the mutation-prone spike protein, they simply switched out the mRNA to one that encodes the full-length nucleocapsid (N) protein. This is the most highly conserved SARS-CoV-2 protein, only rarely changing across variants. The nucleocapsid protein is also the most abundant of the viral proteins, with the membrane and spike proteins coming in a close second. Both of which make it a viable target.
The researchers designed two versions of this vaccine, one that encodes just the full-length nucleocapsid, and another that encodes the full-length nucleocapsid plus the human signaling peptide, CD5 (Figure 2).
FIGURE 2. Schematic representation of the genomes of the two nucleocapsid-based controllable self-amplifying RNA vaccines. Notice that EXG-5005 has the additional CD5 signaling peptide attached to it. FROM: AMANO ET AL. 2022
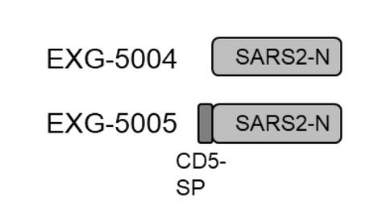
The vaccine without the signaling peptide induced only a weak cellular immune response in mice, with lackluster recruitment of both killer T cells and helper T cells. Increasing the dose had no effect. On the other hand, the vaccine that included the signaling peptide induced a robust T-cell response, suggesting that a signaling peptide is required for the effective stimulation of cellular immunity.
Given their initial success, Amano et al. also designed a vaccine intended to protect against SARS-CoV-2 variants and other betacoronaviruses — SARS-CoV-1, MERS-CoV.
The nucleocapsid proteins of SARS-CoV-1 and SARS-CoV-2 are quite similar, but that of MERS-CoV is only 48% identical to the SARS-CoV-2 nucleocapsid. In order to account for this difference, the researchers fused together the nucleocapsid proteins of SARS-CoV-2 and MERS-CoV, producing a chimera nucleocapsid protein (Figure 3). Again, they also included the CD5 signaling peptide.
Intradermal administration of the vaccine managed to induce a strong T-cell response against both the SARS-CoV-2 and the MERS-CoV nucleocapsid proteins.
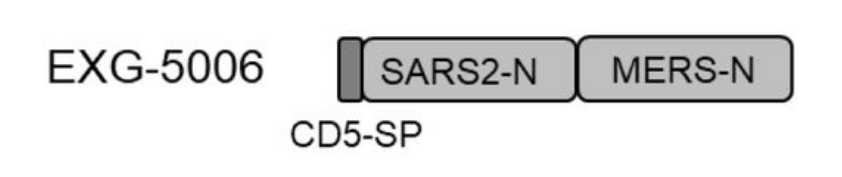
FIGURE 3. A schematic drawing of EXG-5006, encoding a fusion protein of CD5 signal peptide, nucleoprotein of SARS-CoV-2, and a nucleoprotein of MERS-CoV. FROM: AMANO ET AL. 2022
Amano et al. designed one additional vaccine candidate that combined all of the previous elements (Figure 4): SARS-CoV-2 RBD, SARS-CoV-2 nucleocapsid, MERS-CoV RBD, MERS-CoV nucleocapsid, and the CD5 signaling peptide.
As expected, the vaccine managed to stimulate cellular immunity against all of the viral proteins encoded in the construct, indicating its promise as a truly universal pan-betacoronavirus vaccine.
FIGURE 4. A schematic diagram of EXG-5008, encoding a fusion protein of the signal peptide of CD5, the RBD and nucleoprotein of SARS-CoV-2, and the RBD and nucleoprotein of MERS-CoV. FROM: AMANO ET AL. 2022

In Vivo Challenge: Tumor Cells
Next, Amano et al. set out to determine whether the activated CD8+ T cells were successfully destroying infected cells, and by extension, suppressing the spread of the infection. To do so, they engineered tumor cells that expressed the nucleocapsid protein of SARS-CoV-2 and of MERS-CoV. Upon injection, these tumor cells begin to grow and proliferate rapidly, mimicking the dynamics of a viral infection.
The researchers administered an intradermal dose of the temperature-sensitive self-amplifying RNA vaccine encoding the MERS-CoV and SARS-CoV-2 nucleocapsid protein. Three weeks later, they injected the engineered tumor cells.
The tumor cells quickly took hold and proliferated in mice that received only a placebo vaccine. After a period of twenty days, all of them had died from the tumor growth. On the other hand, the growth of tumor cells was largely suppressed in those inoculated with the temperature-sensitive self-amplifying RNA vaccine, with a final survival rate of 40%. The mice that survived were injected with a second round of tumor cells 140 days after initial vaccination; again, tumor growth was quickly suppressed and all of the mice survived.
Takeaways
The most startling result is the fact that unmodified “naked” RNA —without the support of lipid nanoparticles— was able to induce a powerful CD8+ T-cell response. That alone suggests that this approach may prove valuable in adding additional protection to current vaccine strategies.
The experiments also reveal a few weaknesses. Most notably, the absence of a strong humoral immune response. One might argue that the absence of a humoral immune response could be blamed on the use of the receptor binding domain alone, instead of the full-length spike protein. The scientific literature suggests otherwise, with an abundance of studies confirming high neutralizing antibody titers following injection with an recombinant RBD subunit vaccine. The fact that the nucleocapsid protein-based vaccine also failed to induce a neutralizing antibody response illustrates a shortcoming of this approach as a stand-alone vaccine. That may in part be compensated by use of this vaccine as either a primer or a boost to strengthen the CD8+ T-cell responses of conventional mRNA vaccines, or more traditional vaccines that use attenuated or killed viruses.
An additional caveat concerns the temperature-sensitivity of the vaccine; Amano et al. optimized it for skin temperature, which is lower than our core body temperature. This may account, at least in part, for the absence of humoral immunity, since the lymph nodes and germinal centers are located well within the body and function at core body temperature. Germinal centers act as a kind of training camp for B cells. It is where they proliferate and mutate to develop high affinity antibodies against an invading pathogen. If the vaccine antigen is inactive in the germinal centers, then this crucial step of the humoral immune response is omitted.
Future research should aim to test the temperature-sensitive self-amplifying RNA vaccine platform designed by Amano et al. against in vivo viral challenge with SARS-CoV-2, which is notably lacking from this series of experiments. Additionally, it would be very interesting to compare these results with those achieved by conventional, non-temperature-sensitive self-amplifying RNA vaccines and even with those achieved by traditional mRNA vaccines. In some respects, it is surprising that this data was not included in this study.
Read Dr. Haseltine's latest piece with
![]()
